跟着顶刊学分析之单细胞组学结合统计方法解析分布差异

本示例参考Tissue-resident memory CD8 T cell diversity is spatiotemporally imprinted
本文的分析方法和可视化技巧非常值得在单细胞空间转录组研究中借鉴和应用。使用模块设计,将功能分解为多个可复用的函数,全名分析单细胞空间转录组数据,从基因表达到空间关系,再到分组比较。
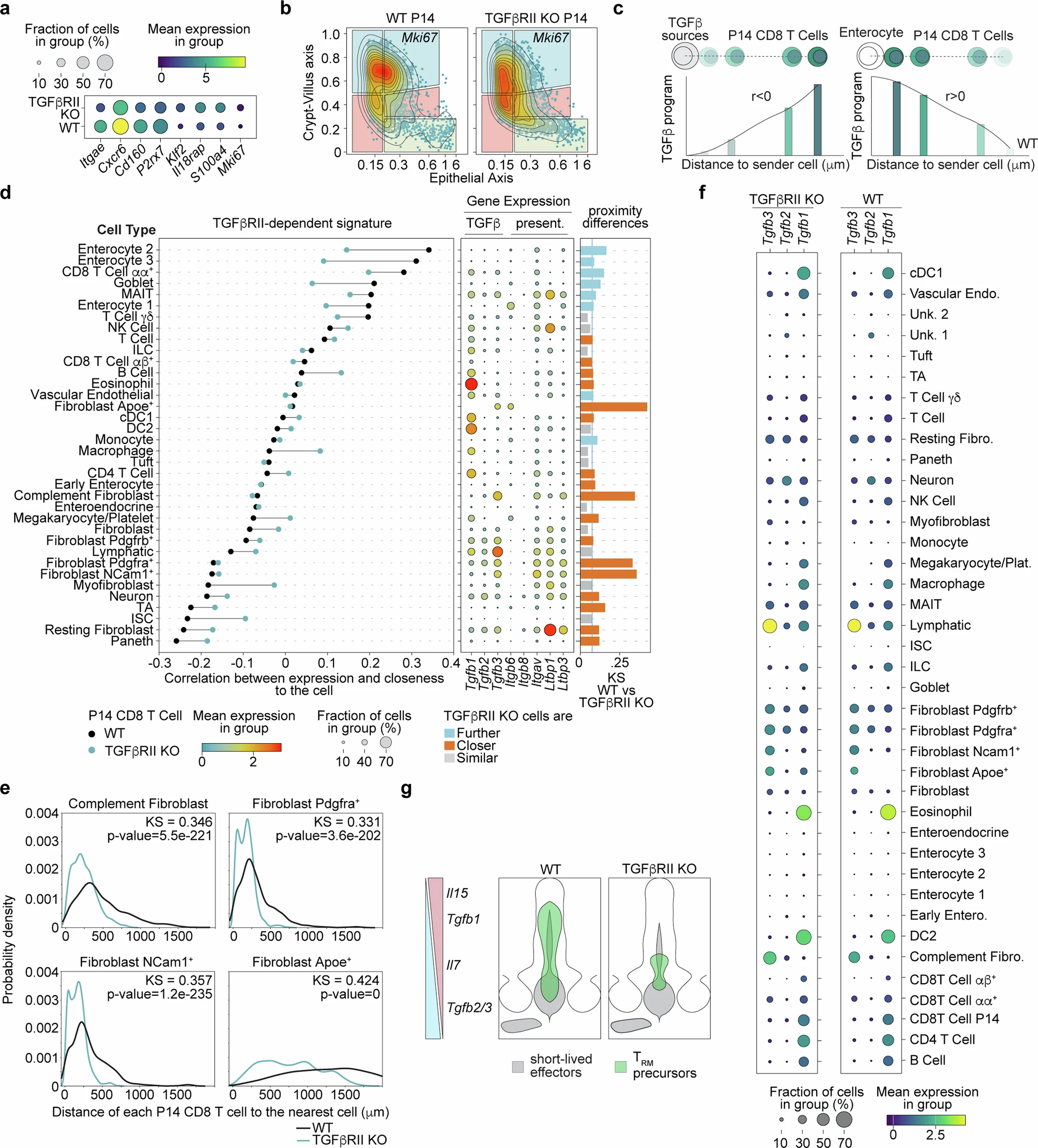
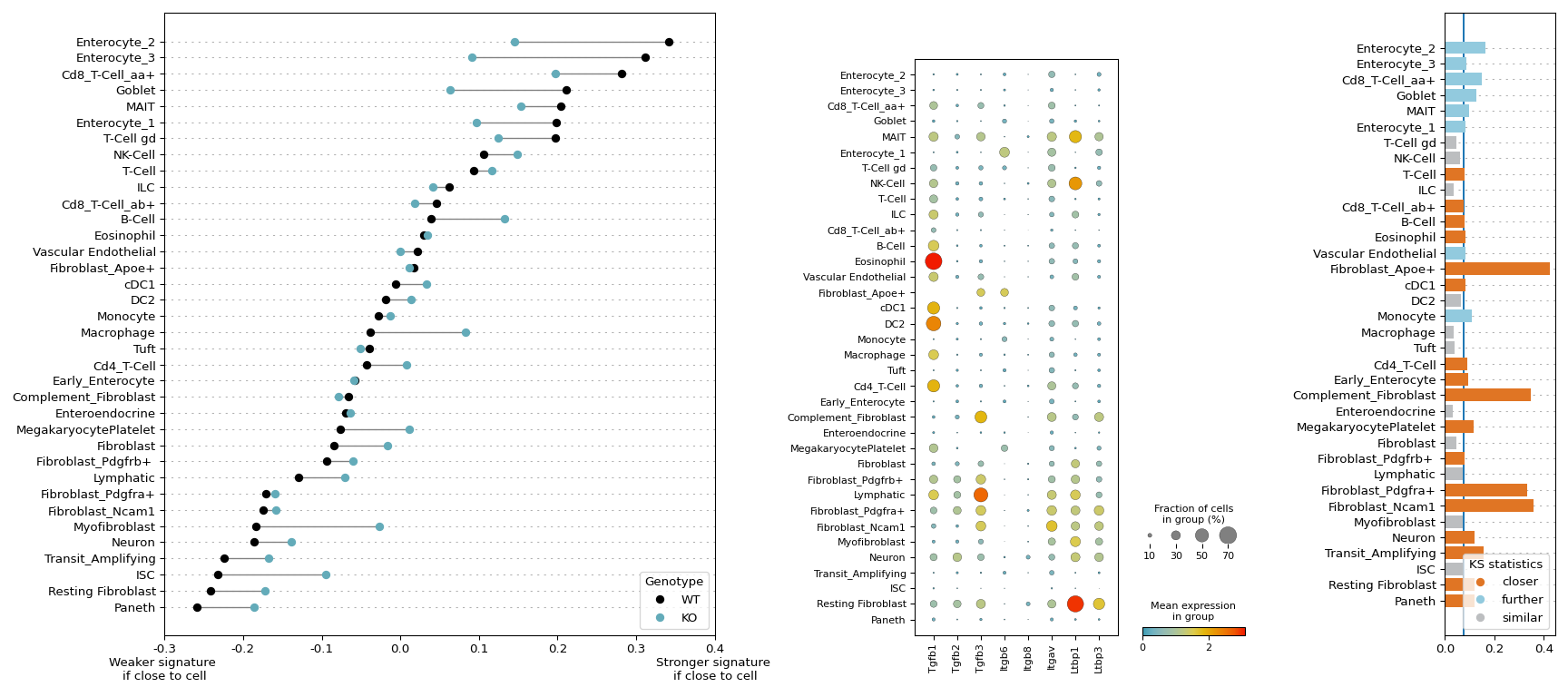
d For every subtype, (left) the correlation coefficients between signature enrichment and P14 cell proximity to the subtype among both WT and TGFβR2 KO P14 CD8 T cells, (middle) the expression of TGFβ isoforms and genes involved in TGFβ presentation in the WT sample, and (right) a non-parametric two-sided Kolmogorov–Smirnov statistic indicating the significance of difference of the distance distributions between P14 CD8 T cells and the corresponding cell type in both WT and TGFβR2 KO. The color of the bars indicates whether P14 CD8 T cells are closer to a given cell type in WT (blue) or TGFβR2 KO (red), and a line indicating effect relevance is positioned at 0.08. Supplemental Table 8 presents the cell counts used in the statistical test for the n = 1 experiment across each condition. e, Comparisons of the distance between WT or TGFβR2 KO P14 cells and selected other cell subtypes. A two-sided Kolmogorov–Smirnov statistic indicates the difference between the WT and KO distributions for each subtype. The plotted lines show the positional density using a 1D kernel density estimate.
图 d 这个复合图展示了 CT8 T 细胞 (P14 亚型)与各细胞亚型的空间互作特征:
- 左图:展示了在野生型(WT)和 TGFβR2 敲除(KO)P14 CD8 T细胞中,某细胞亚型的特征基因评分(DEG)与 P14 细胞距离的 Spearman 相关系数;黑点(WT) vs 蓝点(KO)的偏移表示TGFβR2 缺失改变空间调控模式
- 中图:展示了 WT 样本中 TGFβ 通路相关基因的表达情况
- 右图:P14 CD8 T细胞与各细胞亚型的空间分布差异(KS统计量);橙色条形表示KO 组中 P14 细胞更接近该亚型,蓝色条形表示WT 组中 P14 细胞更接近该亚型,灰色条形表示无显著差异(KS < 0.08)
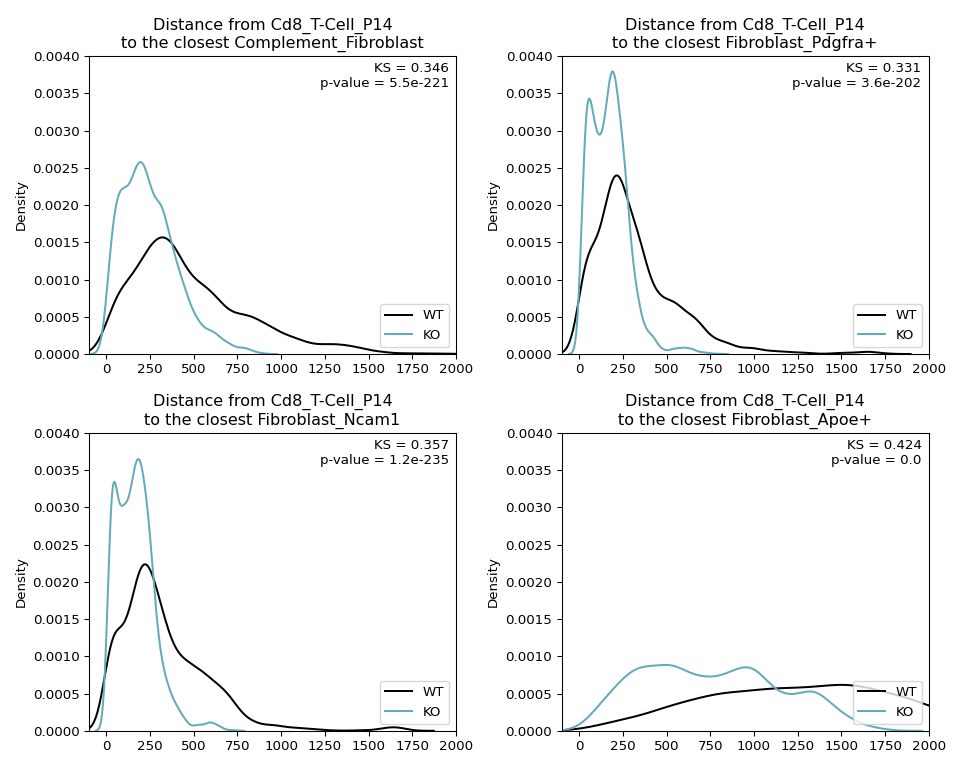
图 e 进一步对关键亚型距离分布做了直接比较;核密度估计曲线展示 WT(黑)与 KO(蓝)中 P14 细胞到目标亚型的距离分布,顶部显示 KS 值与 p 值。
设置运行环境
1 | # 指定 Python 环境 |
导入所需库
1 | import matplotlib |
定义颜色
1 | zissou = [ |
加载数据
1 | adata = sc.read_h5ad("data/tgfb.h5ad") |
数据处理
1 | # 数据标准化处理 |
特征基因评分
1 | # 定义感兴趣的特征基因(DEG:差异表达基因) |
定义核心函数
从数据中提取表达值
1 | # 从数据中提取表达值 |
计算细胞间的空间距离
1 | # 计算细胞间的空间距离 |
分析基因表达与细胞距离的相关性
1 | # 分析基因表达与细胞距离的相关性 |
分组计算距离与表达的相关性
1 | # 分组计算距离与表达的相关性 |
比较两个组间距离分布的KS检验统计量
1 | # 比较两个组间距离分布的KS检验统计量 |
可视化函数
KS 检验结果的条形图
1 | # KS 检验结果的条形图 |
基因表达的气泡热图
1 | def make_expression_plot( |
相关性分析的哑铃图
1 | def make_dot_plot(df, order, ax): |
绘图:哑铃图+气泡热图+条形图
计算 CD8+T 细胞 (P14) 的 DEG 评分与到其他细胞距离的相关性
1 | df = get_batchwise_correlation_between_distance_and_expression( |
计算 KS 统计量
1 | df_ks = get_ks_statistics(adata, "Cd8_T-Cell_P14") |
可视化
1 | # 确定亚型显示顺序 |
绘图:距离分布核密度图
可视化 CD8 T细胞 (P14) 与不同成纤维细胞亚型间的距离分布差异
核心函数
1 | def plot_histogram( |
可视化
1 | # 绘制多个成纤维细胞亚型的距离分布 |
版本信息
1 | import sys |
- Titre: 跟着顶刊学分析之单细胞组学结合统计方法解析分布差异
- Auteur: Xing Abao
- Créé à : 2025-05-12 16:42:18
- Mis à jour à : 2025-10-22 12:15:26
- Lien: https://bioinformatics.vip/2025/05/12/SCI/250512_sci_spatial_scrna/
- Licence: Cette œuvre est sous licence CC BY-NC-SA 4.0.